ABSTRACT
Cryo-electron microscopy (cryo-EM) has become a tool of fundamental importance in structural biology, helping us understand the basic building blocks of life. The algorithmic challenge of cryo-EM is to jointly estimate the unknown 3D poses and the 3D electron scattering potential of a biomolecule from millions of extremely noisy 2D images. Existing reconstruction algorithms, however, cannot easily keep pace with the rapidly growing size of cryo-EM datasets due to their high computational and memory cost. We introduce cryoAI, an ab initio reconstruction algorithm for homogeneous conformations that uses direct gradient-based optimization of particle poses and the electron scattering potential from single-particle cryo-EM data. CryoAI combines a learned encoder that predicts the poses of each particle image with a physics-based decoder to aggregate each particle image into an implicit representation of the scattering potential volume. This volume is stored in the Fourier domain for computational efficiency and leverages a modern coordinate network architecture for memory efficiency. Combined with a symmetrized loss function, this framework achieves results of a quality on par with state-of-the-art cryo-EM solvers for both simulated and experimental data, one order of magnitude faster for large datasets and with significantly lower memory requirements than existing methods.
FILES
CITATION
A. Levy*, F. Poitevin*, J. Martel*, Y. Nashed, A. Peck, N. Miolane, D. Ratner, M. Dunne, G. Wetzstein, CryoAI: Amortized Inference of Poses for Ab Initio Reconstruction of 3D Molecular Volumes from Real Cryo-EM Images, European Conference on Computer Vision (ECCV) 2022
@inproceedings{Levy2022,
author = {A. Levy and F. Poitevin and J. Martel and Y. Nashed and A. Peck and N. Miolane and D. Ratner and M. Dunne and G. Wetzstein},
title = {{CryoAI: Amortized Inference of Poses for Ab Initio Reconstruction of 3D Molecular Volumes from Real Cryo-EM Images}},
booktitle = {European Conference on Computer Vision (ECCV)},
year = {2022}
}
CryoAI Overview
|
|
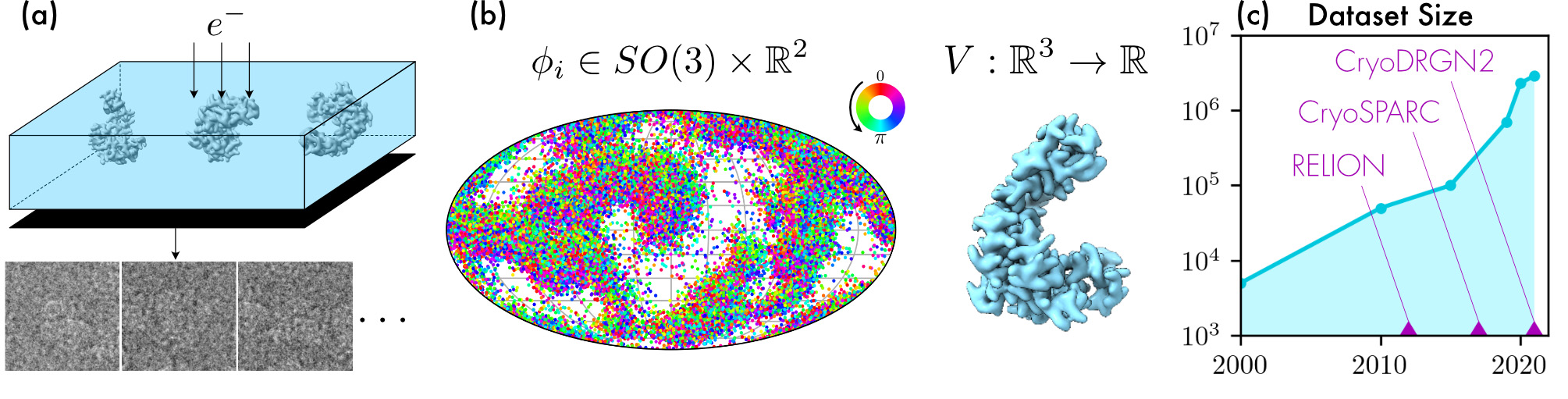
| (a) (Top) Illustration of a cryo-EM experiment. Molecules are frozen in a random orientation and their electron scattering potential (i.e., volume) V interacts with an electron beam imaged on a detector. (Bottom) Noisy projections (i.e., particles) of V selected from the full micrograph measured by the detector. (b) Output of a reconstruction algorithm: poses and volume V. Each pose is characterized by a rotation in SO(3) (hue represents in-plane rotation) and a translation in R2 (not shown). An equipotential surface of V is shown on the right. (c) Evolution of the maximum number of images collected in one day and established and emerging state-of-the-art reconstruction methods. |
|
|
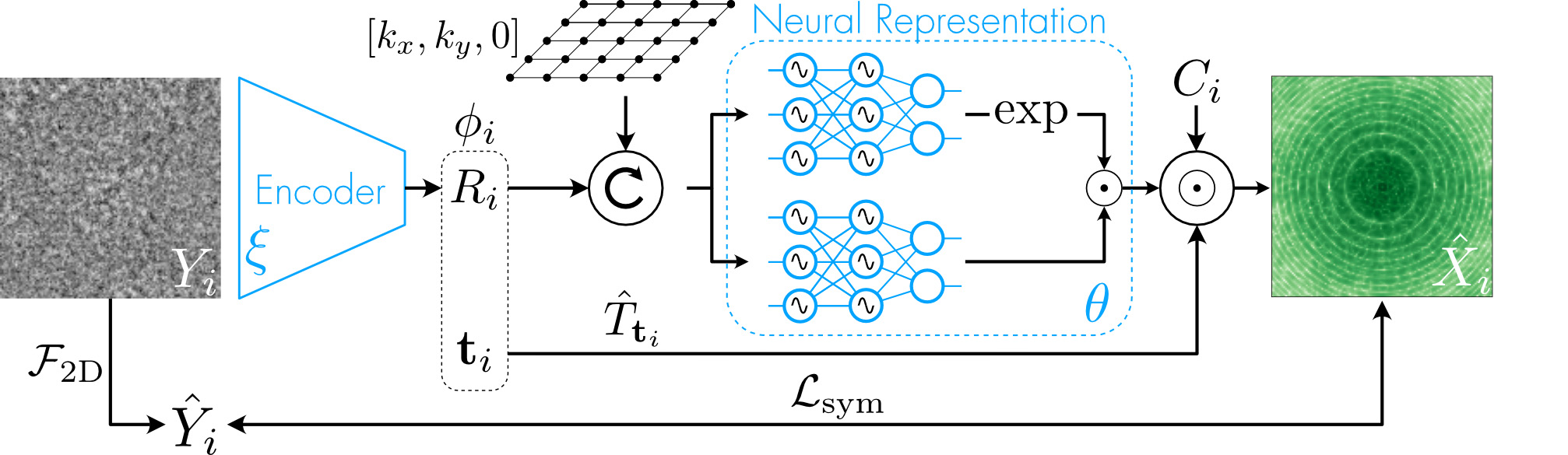
| Overview of our pipeline. The encoder learns to map images to their associated pose. The matrix R rotates a slice of 3D coordinates in Fourier space. The coordinates are fed into a neural representation of the volume. The output is multiplied by the contrast transfer function (CTF) and the translation operator T to build the Fourier representation of the volume. Differentiable parameters are represented in blue. |
|
|
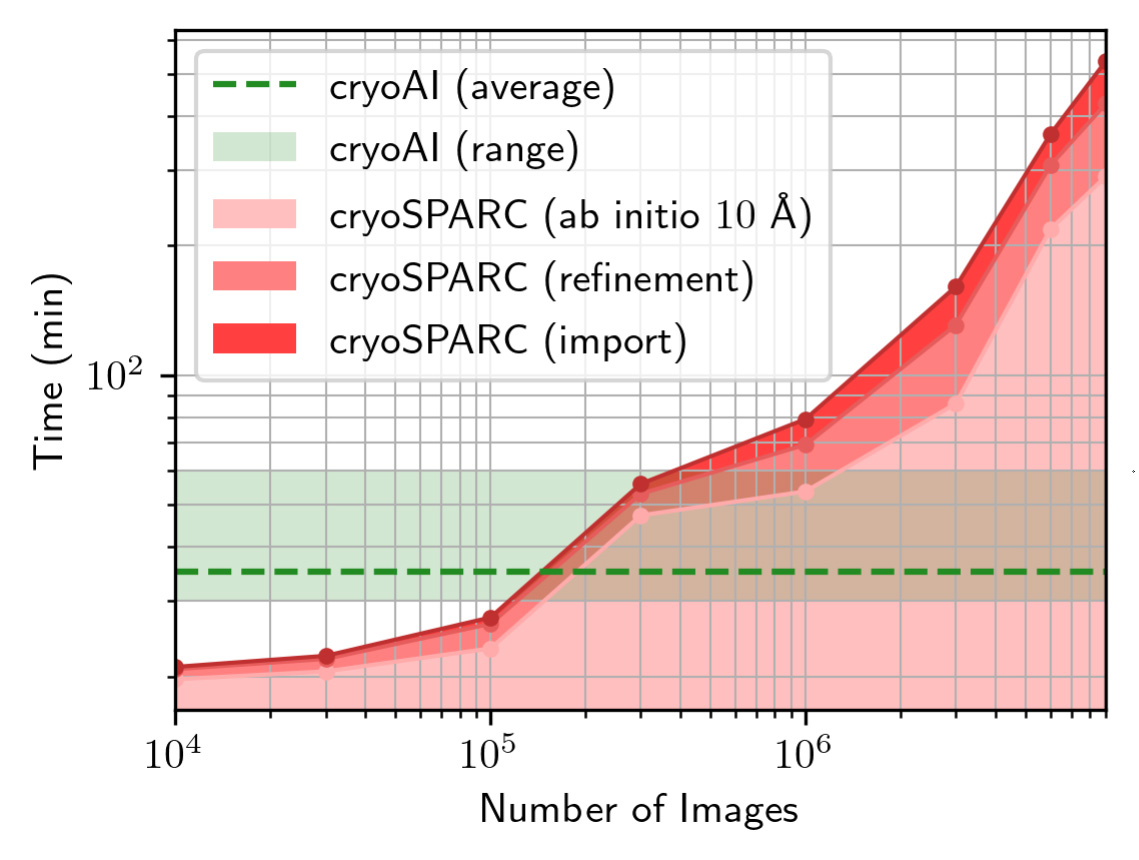
| Time to reach 10 Å of resolution with CryoAI (range and average over 5 runs per datapoint) and cryoSPARC vs. number of images in the simulated 80S dataset. As opposed to existing tools, which scale poorly with increasing dataset sizes, cryoAI provides a nearly constant runtime that amortizes with the size of the dataset, i.e., the number of cryo-EM particle images. |